



Posted by Gregory J Block MSc PhD on May 23, 2016

Summary of Proceedings of the Friends of FSH Research: Research Summit
Abbi L. Engel PhD1, Karla Blonsky MBA2., and Gregory Block PhD1
1. Friends of FSH Research 2. BioBasix Consulting
Correspondence: greg@fshfriends.org
Introduction:
Friends of FSH Research (Friends) is a Seattle-based non-profit whose mission is to accelerate the discoveries leading to a treatment or cure for FSH Muscular Dystrophy (FSHD). The organization has funded over $3 million towards scientific research since 2005 and has helped spearhead many of the major advances in the field. On February 29 – March 1, 2016, Friends held an invitation-only, International Research Summit to identify priority funding areas to accelerate the development of effective therapies for FSH muscular dystrophy (FSHD). The sessions were structured to gather input from academic and clinical researchers, industry personnel, regulators, and other foundations about how best to remove bottlenecks that can slow translation of recent research advances into therapeutic benefit for patients.
Meeting Introduction
Speaker: Gregory Block, PhD Scientific Director Friends of FSH Research
Dr. Block proposed a question to the audience to frame the purpose of the meeting: Is FSHD Treatable? From a basic science perspective, the breakthroughs achieved over the last few years have solidified that FSHD is theoretically treatable. By stopping the FSHD-causing protein, DUX4, the disease should be halted in its tracks. This uncovers the next complicated layer in the puzzle of FSHD. If a drug were designed that reached its target, how would we know that it worked? Drug development is an intricate dance between academic research, industrial rigor, and regulatory oversight. While we continue to support the quest to find a treatment or cure for FSHD, now is the time to engage all the key stakeholders to learn what barriers are holding us back from success.
Session 1: FSHD in 2016
Speaker: Rabi Tawil, MD University of Rochester Medical Center
A summary of the clinical presentation and progression of FSH was provided, highlighting the challenges presented by the extreme variability seen. An overview was presented of the Rochester natural history study that tracks 700 patients, roughly half of whom have confirmed genetic diagnoses. The study has existed for 15 years, and includes efforts patient-reported outcome instrument development, a tissue repository, and clinical trial enablement (e.g. identification of therapeutic intervention windows, power analysis for trial design). Key challenges highlighted included patient recruitment and retention, low rates of genetic testing, and sustainability of funding for the study. (http://onlinelibrary.wiley.com/doi/10.1002/mus.23949/abstract).
Discussions about outcome measures focused on pragmatic considerations for clinical trials:
- Clinical stage of presentation: late stage measures such as wheelchair use are likely not useful since too much damage will have occurred for therapies to have much impact
- Prevalence: comorbidities like retinal damage or hearing loss appear in too small a subset of the patient population to be useful in a trial setting
- Complexity of the muscle function measures: while the six-minute walk test correlated with strength, more complex measures (such as the timed "up & go" test and combined muscle scores) provided a more complete measure of muscle function and progression
- FSHD contribution to mortality: this endpoint has not been well characterized in FSHD. Suggestions included potentially tracking respiratory function or determining a way to follow up with family members.
- Patient-reported outcome (PRO) measures: PROs were seen as critical to understanding patient priorities and therefore to obtaining regulatory acceptance of a proposed trial design.
Speaker: Silvère van der Maarel, PhD of Leiden University Medical Center
Over the last 10 years, agreement has been reached about the role of DUX4 in FSHD; however, the molecule and genetic contributors to the disease state are not sufficiently well characterized. Discussions focused on current efforts to understand how these modifiers play a role in the variability of the disease
- SMCHD1: Although mutations in SMCHD1 are present in less than 10% of the FSHD population, overexpression may suppress DUX4 activity in all FSHD muscles.
- Unaffected family members: A subset of family members have altered repeat size, but with no presentation of FSHD symptoms. Comparison of genetic and epigenetic factors from these individuals has led to biomarkers predictive of presentation/suppression of the disease symptoms. New genes that cause FSHD in very small subsets of patients have been identified.
- Methylation status: Methylation of the D4Z4 repeat array partially predicts FSHD severity. Any genes that influence chromatin condensation could be used to evaluate disease severity or stage. For example, co-inheritance of a mutation in SMCHD1 with a pathogenic D4Z4 array results in a very severe phenotype in FSHD patients.
Implications for therapeutic development and Friends funding:
Deconvolution of the complexity of FSHD will be essential to ensure that [i] therapeutic efficacy can be detected and [ii] proper patient cohorts can be selected for clinical trials.
High priority opportunities included:
- Fund genetic testing for the undiagnosed individuals of the US registry
- Support validation studies of functional outcome measures in the FSHD population
- Evaluation of composite scores and patient-reported outcome instruments
- Identification and validation of biomarkers to establish a link with disease state (severity, rate of progression, muscle group involvement, etc.)
Note: Although these areas urgently require additional investigation before the disease mechanism can truly be understood, drug development and clinical studies are able to proceed based on the current levels of knowledge.
Session 2: Lessons learned in rare disease therapeutic development
Speakers: John Porter (Consultant; Former Parent Project, NIH), Bruce Wentworth (Sarepta), Laura Hagerty (MDA)
Therapeutic development in other rare diseases was discussed, highlighting lessons of relevance for FSHD where the field can act now to avoid downstream delays and failures:
- Disease models: It is imperative that disease models are predictive of the disease state in humans and that changes in assays/animals can be quantifiably linked to meaningful improvement in patients. The key questions for FSHD will be:
- What reduction of DUX4 is required for therapeutic benefit in humans?
- In animal models of the disease, how much can levels be knocked down before they become problematic?
- Efficacy v. investigational studies: While some preclinical models are best suited for efficacy studies, others might be useful for pharmacodynamics, pharmacokinetics, or bioavailability. Similarly, a “tool” compound identified in a disease model can be useful to determine mechanism or to interrogate the biology, even though it may not be a candidate to carry forward into downstream development.
- Assay and screening efforts: Discussions focused on choosing the optimal assay readout, using proper controls, having well-powered groups for statistical analysis and when possible, using blinded and randomized study structures (see NINDS Rigor Guidelines; Guidance on Bioanalytical Method Validation).
- Research QC: Preclinical studies should be structured with the same focus on rigor, validation, and reproducibility as is seen in clinical studies. Examples cited included:
- Reproducibility issues in Pompe disease, where the candidate that was identified to be most efficacious depended on the experiment conducted.
- In ALS, repetition of key experiments with clinical candidates at a different site were unable to demonstrate the same efficacy.
- With biologics, issues have been seen as larger quantity production resulted in decreased activity, highlighting the need for constant product QC and re-evaluation of efficacy during scale up.
In addition, informing early research about the downstream requirements can have enormous benefit in terms of more robust data packages about compounds, earlier go/no go decisions, and faster regulatory reviews in IND-enabling stages.
Implications for therapeutic development and Friends funding:
Because the cost of a clinical trial is roughly $5 million per day, improvements to early stages of research can have tremendous return on investment. Funding by Friends should therefore be tied to metrics that ensure stringent that quality rigor, validation, and reproducibility requirements are met.
High priority opportunities included:
- Enhanced characterization of current animal models
- Side-by-side experimental strategies to evaluate the utility/reproducibility of the tools used to measure efficacy
- Funding of third party validation of key results before advancing a lead compound to decrease the risk of taking ineffective drugs into the clinic.
Session 3: Small molecule therapies
Speakers: Anton Simeonov, PhD (NCATS/NIH), Fran Sverdrup, PhD (St. Louis University), David Dasberg (Facio Therapies), Jamshid Arjomand, PhD (Genea Biocells)
Discussions focused on the development of small molecule therapies, including assay development and screening, creating partnerships with industry, and developing drug candidates. An overview was presented of the technical and funding resources at NCATS (National Center for Advancing Translational Sciences), which cover early assay development, uHTS, lead optimization and preclinical support. NCATS offers a 500,000 compound libraries that include known drugs, compounds that probe specific mechanisms, and purified natural products (see Appendix 1). In addition, their ultrahigh throughput screening (uHTS) system has the potential to look at combinatorial effects of more than one compound at a time.
The NIH has also compiled some of the best practices to create the "Manual for Assay Guidelines" based on Eli Lily’s process (www.ncbi.nlm.nih.gov/books/NBK53196/). In addition to technical assistance, the TRND (Therapeutics for Rare and Neglected Diseases) program is also supports preclinical collaboration efforts.
Additional presentations focused on assays currently being developed at industry and academic sites, as well as key considerations that inform screening efforts:
- Genea Biocells described their repository of stem cells available to investigators, including three human ES cell lines registered with the NIH that are provided with a scalable protocol for muscle cell differentiation. Internal screens at Genea have shown that DUX4 expression affects the morphology of the myotubes and can be used as one of the measures in their assay. Genea is also pursuing functional endpoints that measure force and contraction using their cells.
- Facio Therapies described their assay that uses primary FSHD muscle cells to look at DUX4 and SMCHD1 modulation.
- The choice of the cell type and the assay should be determined by the ultimate purpose intended for the screen, and may require trade-offs. For instance, stem cells might be easier to scale up, but differentiated myotubes have less variability and consequently offer better reproducibility for larger scale screens.
Implications for therapeutic development and Friends funding:
It will be critical to conduct future assay development and screens on a scale that will enable comprehensive interrogation of the biology and therapeutic efficacy.
High priority opportunities included:
- Facilitating researcher access to advanced uHTS capabilities and to as many extensive compound libraries as possible. This work will therefore be best handled at centers such as NCATS that have industrial scale capabilities.
- In-depth evaluation of how representative assays are of the disease mechanism/pathology.
- Conclusion reached that funding academic screens is a futile activity.
Session 4: Antisense Oligonucleotide and gene therapy
Speakers: Olivier Danos, PhD (Biogen), Scott Harper, PhD (Ohio State University School of Medicine)
Several biologic strategies could be used to treat FSHD. For antisense oligonucleotides, targets could be DUX4 mRNA degradation or splicing. In gene transfer, shRNA could be used with strategies that alter chromatin modifiers (like SMCHD1 and DMTB3). Lastly, gene editing strategies could also target chromatin modifiers, PAS, or locus repair.
However, clinical development of antisense oligonucleotides and gene therapies poses unique challenges. Discussions focused on several issues that require attention:
- Safety (particularly heart and liver toxicity due to saturation), although it may be more straightforward to prove safety for a knock-down therapy than for other additive therapies.
- Establishment of the connection between therapeutic action on the target and changes in pathophysiology. For instance, if evaluating the knock down of DUX4, a link to the symptomology will be necessary to evaluate if the vector is suitable for therapeutic purposes. Questions were raised regarding the effect of chronic suppression of DUX4 in terms of side effects/adverse events, but consensus was that these issues should not be prohibitive since abnormally low DUX4 expression has been seen in patients without severe problems.
- Dosing may be problematic. The possibility of using direct injection into affected muscles was presented. However, dosing would need to be a one-time treatment to avoid immune activation, unless an immune suppressant is administered along with the vector.
- Scale up of vector manufacturing is non-trivial and will require considerable time, effort, and money.
Implications for therapeutic development and Friends funding:
AON and gene therapies may offer the most direct path to address the disease mechanism and, therefore, may lead to more effective treatment options.
High priority opportunities included:
- Expanded characterization of animal models to evaluate gene therapy and AON therapies.
- Improved understanding of the consequence of DUX4 suppression and disease phenotype.
Day 2: Paradigm shifting opportunities
Session 5: Regulatory discussion
Speakers: Pavel Balabanov, MD/PhD (European Medicines Agency), Allison Morgan (Metis Clinical), Libby Wood (UK FSHD registry curator & project manager; Newcastle University)
Understanding the regulatory perspective on therapeutic development in FSHD informs research at all stages of drug development. In order to identify the optimal therapeutic window in the right patient cohort, the FSHD field will need to be able to predict disease progression during a trial so it can be determined if a drug is having a therapeutically beneficial impact. As well, variability in FSH will pose challenges for regulators to assess the cost/benefit ratio because predicting who will likely benefit from the therapy (I.e. who would have progressed without the drug) is not feasible with the current level of understanding about disease pathophysiology. Futility trials using historical data as control will likely not be sufficient and placebo-controlled designs are expected to be required.
Early engagement of regulators was seen as an important next step for the FSHD field, with mechanisms for discussions available at all development strategies from early therapeutic innovation through later development strategy discussions. It was strongly encouraged to speak with regulatory agencies BEFORE first human studies to address issues that could delay therapeutic development. For example, opportunities to review the utility of proposed endpoints and biomarkers with the EMA in advance of trials were discussed to ensure proper validation of the measures for their use in advance of trials. Additional avenues for researchers to engage with regulatory development groups were highlighted, including the Innovation Task Force (EMA) and the Critical Path Initiative (FDA).
Discussions also reinforced the urgent need for reliable, robust biomarkers and clinical outcomes in FSHD that can be linked with functional improvement that matters to patients. In DMD, dystrophin use as a surrogate endpoint was rejected because no clear link had been established between the levels of the biomarker and the demonstrated, clinically meaningful changes in patients. Because measures must relate to patient priorities, it will be important to take into account both physician and patient perspectives. For instance, clinical assessment may typically employ the 6-minute walk test, but patients are more likely to value the ability to dress themselves. Patient-reported outcomes (PROs) were cited as being highly useful for this purpose. FSHD-Hi criteria are being evaluated for their use in clinical studies in FSHD. Due to patient variability, a battery of tests such as electrical impedance, force generation (oral pressure), axial weakness or upper limb measure adapted for FSHD relevant criteria may be required.
TREAT-NMD (Translational Research in Europe - Assessment and Treatment of Neuromuscular Diseases), a UK-based group provided an overview of their resources to help standardize research and enable clinical trials in neuromuscular disease, including standardized animal model protocols, clinical care standards, outcome measures and tissue repositories. They also have advisory resources such as TACT for developing therapeutics, Project Ethics Council (PEC), training and education programs, and individual project support initiatives. In FSHD, the group also has an initiative to collect a minimal core dataset on patients from registries around the world.
Although there are currently registry efforts in FSHD in several countries, there was consensus that additional efforts should be focused on moving toward harmonization between registries in order to get the most value from the clinical data. Beyond recruiting patients for studies, these registries also assess standards of care and quality of life. These registries can help to determine disease prevalence, explore patient preferences, calculate burden of illness and provide post-marketing surveillance.
Questions related to the best platform to use, what data points should be collected, implementation of global identifiers, and other logistics remain to be solved. As well, while registries can provide useful information, they cannot substitute for comprehensive longitudinal natural history studies that will be required for clinical trial enablement.
Implications for therapeutic development and Friends funding:
Better characterization of the patient population and improved understanding of patient priorities will be critical for clinical trials. Unless this is addressed in the near term, it poses significant risk of delaying therapeutic development.
High priority opportunities included:
- With the FSHD Champions, harmonization of currently funded registries to the greatest extent possible.
- Natural history studies to improve understanding of the heterogeneity of the disease and the variability in its progression.
- Characterization and validation of outcome measures for use in clinical studies, including third party reproducibility studies. Researchers, clinicians, patient advocacy and non-profit groups must work together to identify the tools that will be used for clinical evaluation, and validate them in the FSHD patient population before effective clinical trials can be possible.
Session 6: Modeling the disease
Speakers: Michael Kyba, PhD (University of Minnesota), Robert Bloch, PhD (University of Maryland School of Medicine)
Animal models that accurately recapitulate the disease state in humans will be essential to evaluate lead compounds in efficacy studies. Various models were discussed, with a focus on their relative utility to understand disease mechanism and assess efficacy of therapeutic candidates:
- Several lower animals (e.g. Drosophila, Xenopus, zebra fish) can assess the effect of genetic manipulations on tissue or development, but these organisms may not be sufficiently predictive of the disease state for preclinical efficacy evaluation.
- Most of the current models were seen as appropriate only for secondary evaluation of drug candidates. For example, the models may be useful for PK/PD evaluation, determining the importance of satellite cells (and DUX4 in those cells), or safety studies that provide support and de-risk a project in pre-clinical development.
- Implantation of human muscle into mouse models provides an interesting mechanism for direct testing in human tissue. However, the disadvantages are that there is significant variability in yield, the process is extremely time and labor intensive, the longevity of the graft and mouse is short compared to time course of FSHD, the graft doesn’t recapitulate the development stages or rostrocaudal gradient, and some of aspects of the disease remain to be characterized for their representativeness of the human state (i.e. nerve, immune, endocrine, or connective tissue involvement).
Implications for therapeutic development and Friends funding:
Animal models used for efficacy testing used must be matched to the specific therapy, such that there is a well-characterized link between the drug’s specific target or molecular mechanism and the pathogenic pathway.
High priority opportunities included:
- Characterization of current animal models to ensure that they can be used to evaluate therapies with sufficient confidence.
- Supporting collaborations between researchers and industry.
- Support of external validation studies.
Session 7: Emerging strategies to track disease state and progression
Speakers: Joel Chamberlain, PhD (University of Washington), Stephen Tapscott, MD/PhD (Fred Hutchinson Cancer Research Center)
The importance of biomarkers in a highly variable disease such as FSHD cannot be overstated. Without markers that are predictive of disease state and progression, it will be impossible to identify the best cohorts to be used in clinical trials. Without early molecular or other indicators of efficacy that can inform dosing, trials will be problematic.
Discussion of possibly biomarkers for FSHD status, severity and progression included:
- DUX4.
- Immunological markers.
- The role of satellite cells.
- MRI to assess the tissue changes in FSHD progression. This may provide a means to evaluate progression from healthy muscle tissue to the fatty infiltrated disease-affected muscle. Currently, MRI and needle biopsy are being used along with physical examination and functional assessment to correlate the status of cells/tissue to disease progression.
- Tissue changes in human biopsies and mouse muscle can be compared via histology and fluorescent staining to demonstrate correlation with the disease phenotype.
Assessment of biomarkers in the animal models had previously been discussed, including assessment of DUX4 production via muscle specific promotor or non-constitutive promotors, assessing the role of polyA or methylation in different species, and the development of murine phenotypes that may or may not be related to the pathology of DUX4 expression.
Implications for therapeutic development and Friends funding:
Animal models used for efficacy testing used must be matched to the specific therapy, such that there is a well-characterized link between the drug’s specific target or molecular mechanism and the pathogenic pathway.
High priority opportunities included:
- Validate the link between the biomarkers and functional outcome measures in patients.
- Include biomarker evaluation in clinical assessment of patients.
- Supporting collaborations between researchers and industry.
- Co-funding larger projects with other FSHD Champions organizations, Muscular Dystrophy Association (MDA), or the Wellstone center.
Session 8: Strategic Planning
This session included discussion with all attendees about funding priorities for Friends (added to each session above) and brief introduction of other opportunities at the Muscular Dystrophy Association (MDA) by Laura Hagerty and June Kinoshita of FSH Society. Laura highlighted opportunities for researchers (training and research grants) and career transition awards through the MDA. Another key grant opportunity is the MDA venture philanthropy award for translational research projects to build infrastructure and bring new ideas or new techniques for research and development projects. The MDA is looking for more proposals in FSHD and has a range of opportunities to support larger/longer running projects.
Overall, the Summit met the goals of Friends of FSH Research:
- engaging all stakeholders and get perspective of industry and regulators on the status of the FSHD research field
- introduce new industry representatives to opportunities in FSHD research & develop relationships to advance drug development
- identify priorities (like expanding clinical research by establishing Seattle clinical center of excellence, improving patient reported outcomes and improving US registry and/or global harmonized registry) that we can begin to shape our funding strategy
- build off of existing work and establish collaborations with institutions (like Wellstone Center, MDA, NCATS) to leverage resources and make donor dollars go further
Appendix 1: Additional information from Anton Simeonov, NCATS
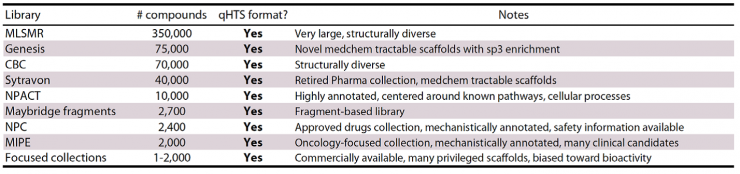
Screening libraries at NCGC: Diverse, curated libraries (+550,000 compounds)
Appendix 2: Additional Information from John Porter:
NINDS “Rigor” Guidelines:

Rationale for Making Go/No-Go Decisions for Clinical Trials
- Is MOA/target sufficiently understood and likely to be disease modulating?
- Is your drug sufficiently understood?
- Degree to which the preclinical model mirrors the human disease (PD or phenotype)?
- Translatability of PK/bioavailability data?
- Robustness of preclinical study design and drug effect?
- Understanding of the patient population
- # and distribution
- Natural history
- PD biomarker(s) that drug is doing what’s intended in the trial
- Clinically meaningful endpoint(s) appropriate for trials
Connect with us on social media